
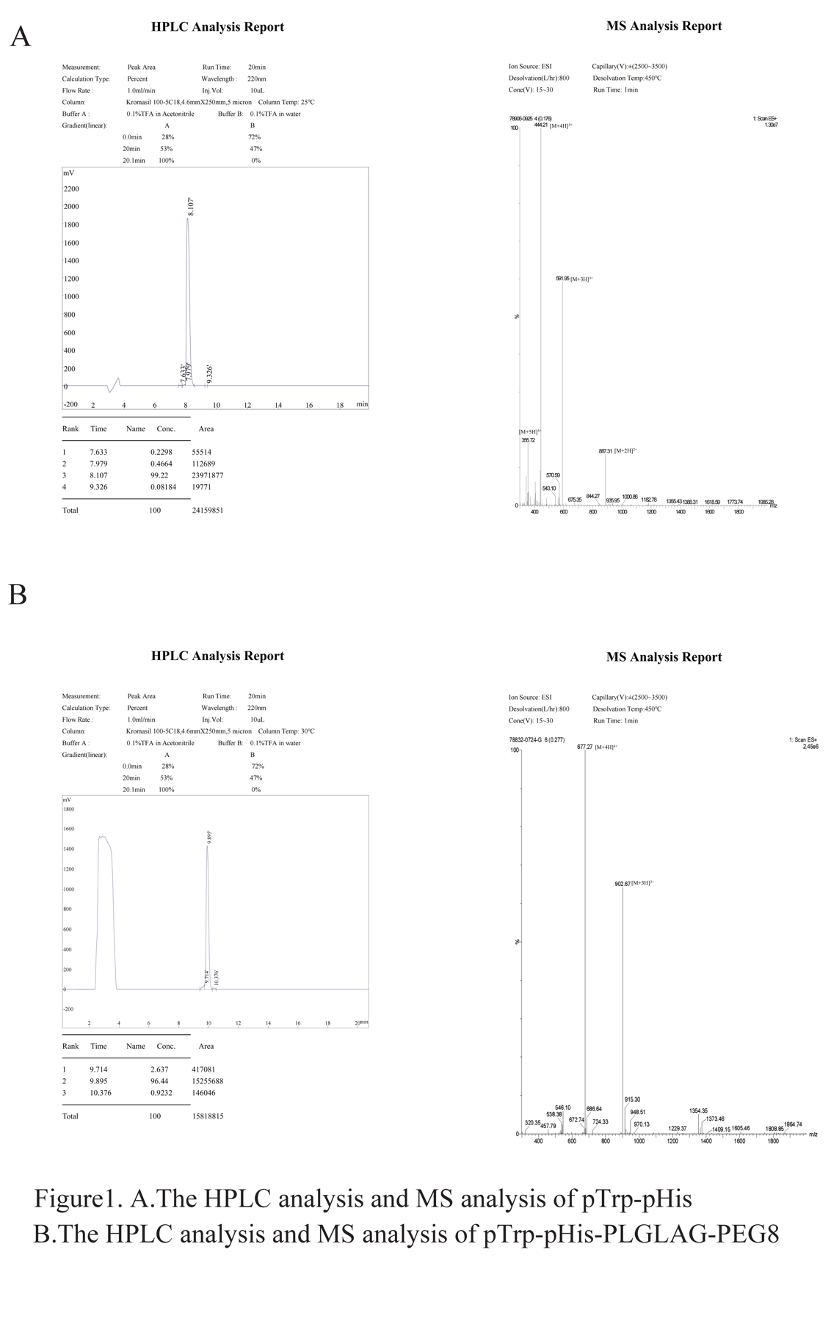
1.1 Synthesis and Characterization of pTrp-pHis and pTrp-pHis-PLGLAG-PEG8
The designed peptide comprises a hydrophobic core of poly(tryptophan)-poly(histidine) (pTrp-pHis) and a hydrophilic chain of polyethylene glycol (PEG-8) linked by a peptide (PLGLAG). The contracted peptide synthesis company provided analytical reports on the purity of pTrp-pHis and pTrp-pHis-PLGLAG-PEG8. Mass Spectrometry (MS) and High-Performance Liquid Chromatography (HPLC) analyses showed that the purity was 99.22% (Figure 1, A) and 96.44% (Figure 1, B), respectively.
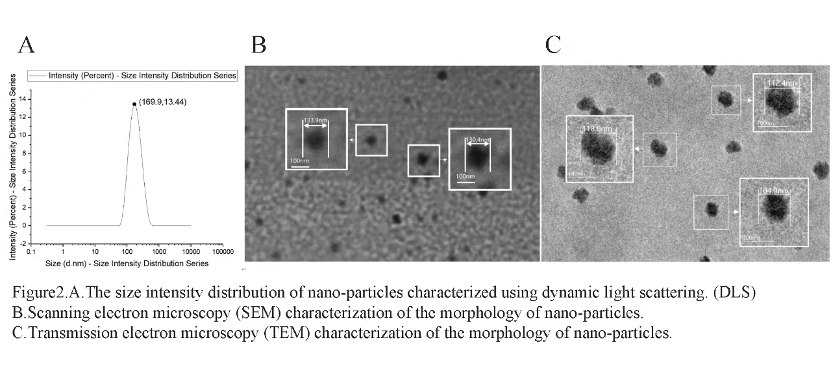
1.2.1 Preparation and Characterization of Polymeric Micelles
The self-assembled nano-particles are around 100 nm in size, as shown by data analysis from Dynamic Light Scattering (DLS) (Figure 2, A), Scanning Electron Microscopy (SEM) (Figure 2, B), and Transmission Electron Microscopy (TEM) (Figure 2, C). However, the size measurements from DLS are larger than those from SEM and TEM. This is because DLS measures the hydrodynamic diameter of nano-particles in solution, which is slightly larger than the physical diameter measured by electron microscopy.
1.2.2 Exploration of Peptide Preparation Conditions
Based on the observation of the Zeta-Potential distribution of pH 7.4 and pH 6.5 solution samples, it can be concluded that the pH 7.4 sample has a larger absolute Zeta-Potential value, indicating that the nanoparticles in the pH 7.4 solution are more stable. This also suggests that under the acidic pH conditions of the tumor microenvironment, the protonation of polyhistidine carrying positive charges will induce the collapse of nanoaggregate structures.
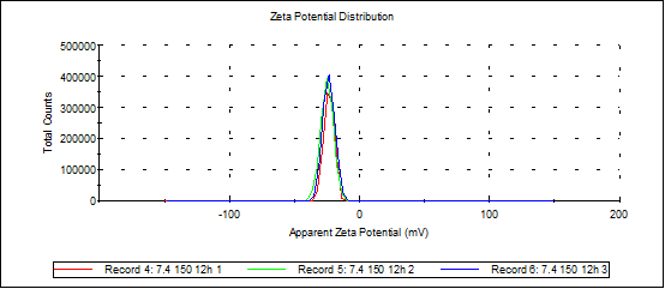
Zeta-potential of sample at pH7.4
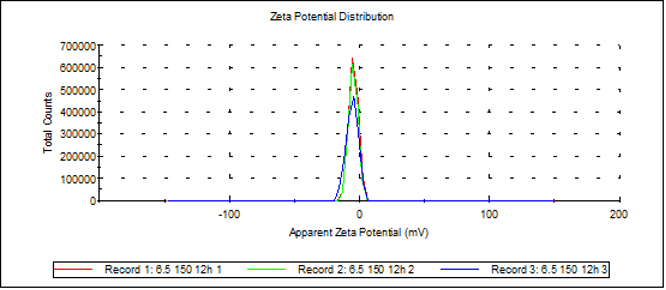
Zeta-potential of sample at pH6.5
In five different concentration solutions, it was observed through DLS particle size distribution plots that when the concentration was within the range of 28.2 to 84.6 μmol/mL, the peak particle size of the assemblies gradually changed from 28 nm to 120 nm. Further observations revealed that when the concentration exceeded this range, the particle size even increased to 1000 nm. This indicates that the optimal assembly range is approximately 84.6 μmol/mL.
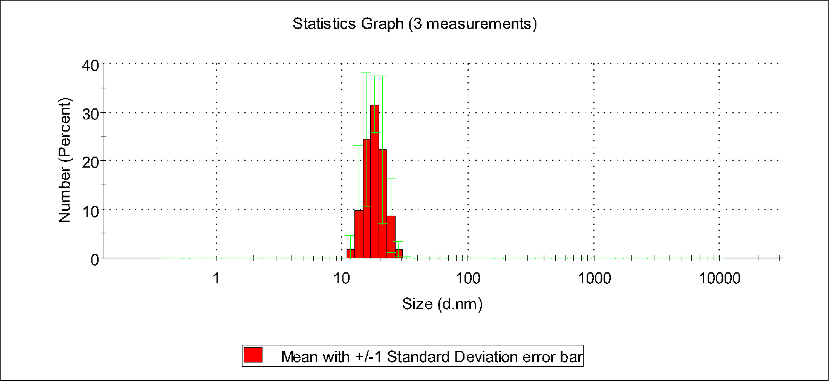
Number stats graph of PBS, pTrp-pHis-PLGLAG-PEG8(12h 0.028 mM pH7.4)
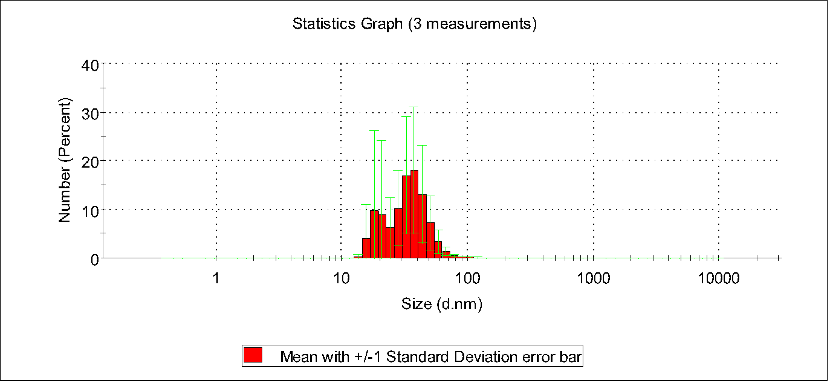
Number stats graph of PBS, pTrp-pHis-PLGLAG-PEG8(12h 0.056 mM pH7.4)
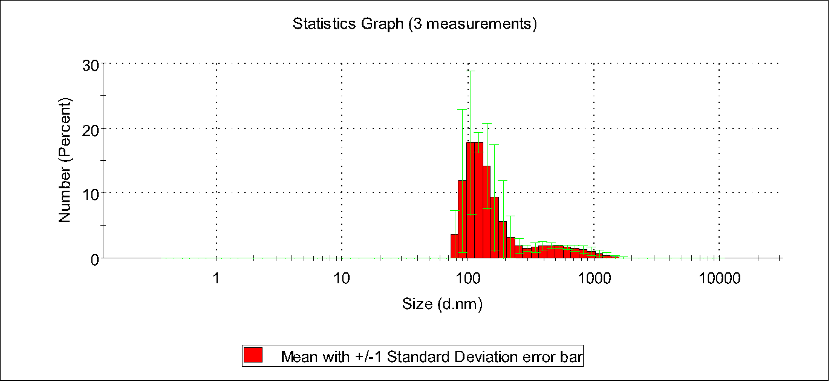
Number stats graph of PBS, pTrp-pHis-PLGLAG-PEG8(12h 0.085 mM pH7.4)
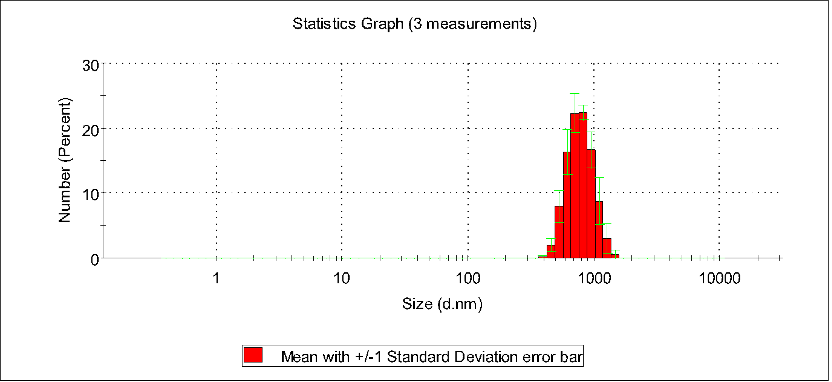
Number stats graph of PBS, pTrp-pHis-PLGLAG-PEG8 (12h 0.113 mM pH7.4)
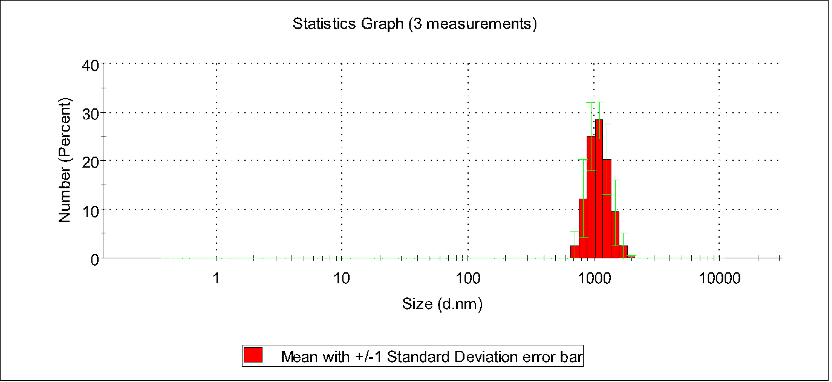
Number stats graph of PBS,pTrp-pHis-PLGLAG-PEG8(12h 0.141 mM pH7.4)
1.3.1 MD simulation of polypeptides monomers aggregation into nanoparticles
The solvent-accessible surface area (SASA) provides an estimate of the number of residues located on the surface regions of the polypeptides, as well as those buried within the hydrophobic core. As depicted in Figure 1, a decrease in SASA values was observed over the 50ns Molecular Dynamics (MD) simulation. This finding suggested that the polypeptides underwent self-assembly during this period. From 50-200ns, the SASA value remained stable, indicating the formation of a stable nanoparticle.
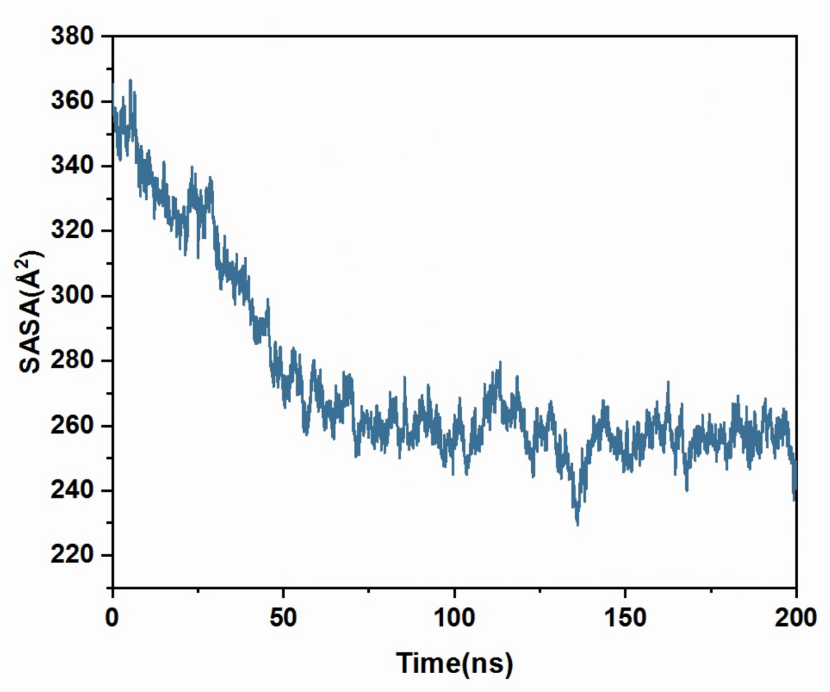
Figure 1. The temporal evolution of the SASA value.
Figure 2 presented the number of hydrogen bonds formed between polypeptides. An increase in this number over the 200ns period suggested that hydrogen bonds play a vital role in nanoparticle self-assembly.
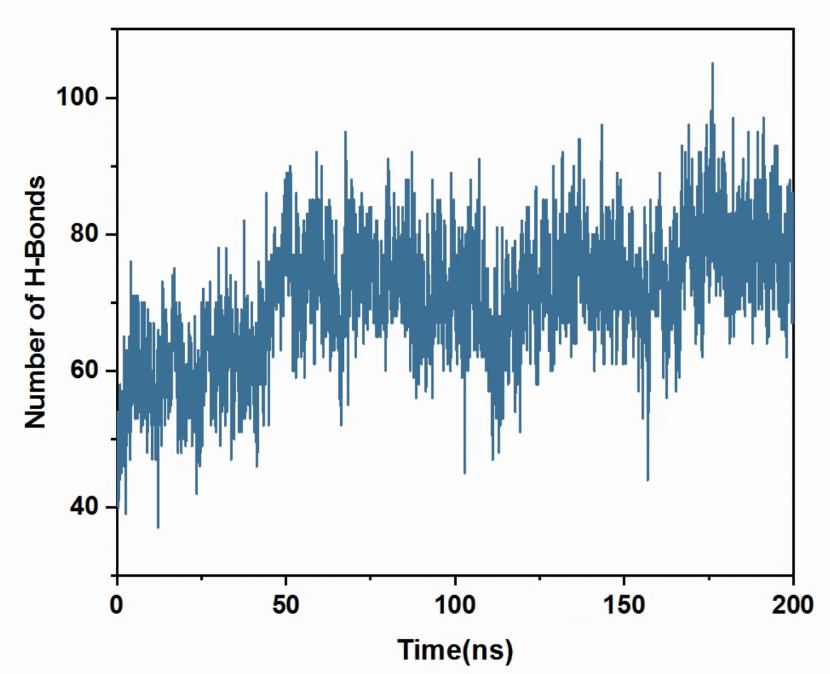
Figure 2. The temporal evolution of the number of hydrogen bonds.
Then we fetched a representative conformation of formed nanoparticle to investigate interaction contributions. As indicated in Figure 3-1, Pi-Pi stacking, Pi-Pi T-shaped, and conventional hydrogen bonds significantly contribute to polypeptides combination. These forces could also potentially serve as the driving forces behind self-assembly.
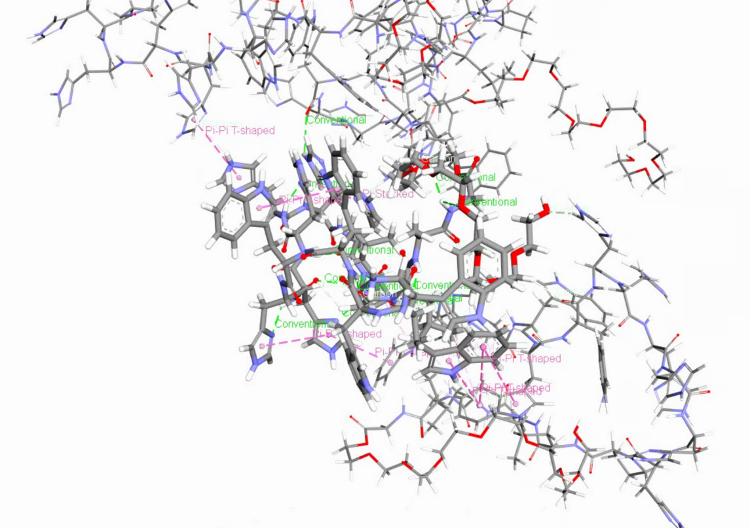
Figure 3-1. interaction contributions of peptides inside nanoparticle.
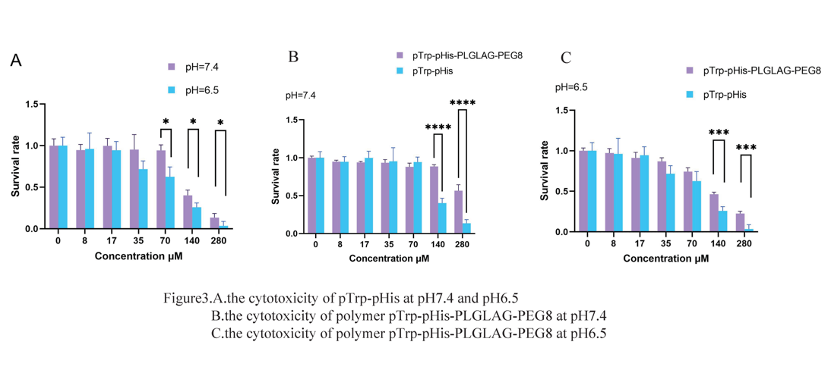
1.3.2 Cytotoxicity of pTrp-pHis under different pH conditions
To assess the cytotoxicity of pTrp-pHis, we performed a CCK assay on HepG2 cells under different pH conditions (6.5 or 7.4). As shown in Figure 3, A, pTrp-pHis had no significant effect on cell viability at low concentrations (0 to 70 μM) after 6 h of incubation at pH 7.4. However, when the concentration increased to 140 μM, the cell viability dropped to about 40%. At pH 6.5, pTrp-pHis reduced cell viability markedly at concentrations from 35 to 280 μM. At the highest concentration of 280 μM, nearly all cells were dead.
The cell viability at pH 6.5 was consistently lower than that at pH 7.4 for the same peptide concentration, and the difference was statistically significant for concentrations above 71 μM. This indicates that an acidic environment enhances the toxicity of pTrp-pHis (Figure 3, A). Therefore, acidic conditions that induce tumor cell death or inhibit tumor growth may potentiate the therapeutic effects of pTrp-pHis.
1.3.3 Cytotoxicity of polymer pTrp-pHis-PLGLAG-PEG8 under different pH conditions
We also evaluated the cytotoxicity of polymer pTrp-pHis-PLGLAG-PEG8 using a CCK assay on HepG2 cells under different pH conditions (6.5 or 7.4). Figure 3, B shows that polymer pTrp-pHis-PLGLAG-PEG8 had no significant effect on cell viability at concentration of 280 μM, cells maintained about 60% viability. At pH 6.5, polymer pTrp-pHis-PLGLAG-PEG8-treated cells exhibited significantly higher cell viability than pTrp-pHis-treated cells at the same concentration (Figure 3, C). This suggests that the shielding of PEG reduces the toxicity of pTrp-pHis considerably. It also implies that polymer pTrp-pHis-PLGLAG-PEG8 has good biocompatibility and is suitable for use.
Amphiphilic molecules can kill tumor cells, but their amphiphilic structure also causes high toxicity to normal tissues. Therefore, the key to applying plasma membrane rupture (PMR) molecules is to achieve high selectivity for target cells, while minimizing their toxicity to normal tissue cells.
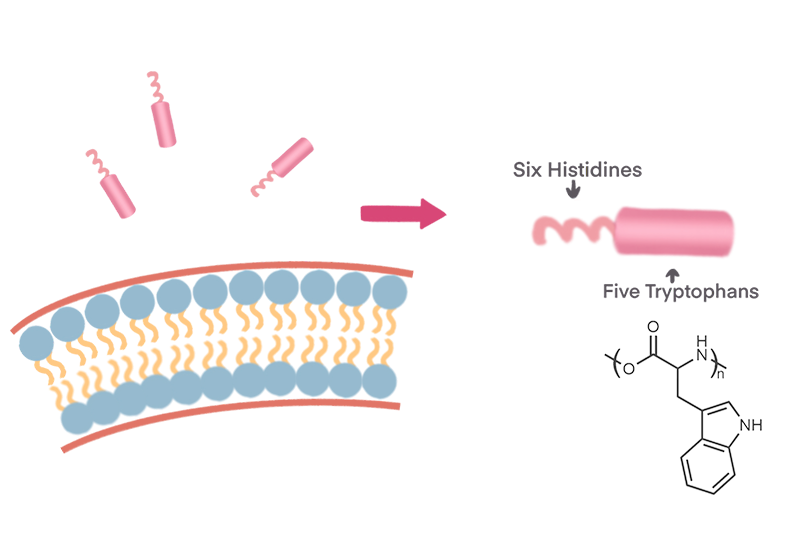
When we choose peptides to design amphiphilic molecules, the function and efficacy of the hydrophobic moiety are critical. Amino acids can generally be classified into polar and non-polar amino acids, among which non-polar amino acids include two special aromatic amino acids: phenylalanine and tryptophan. Studies have confirmed that in the hydrophobic moiety, compared with alkyl groups (such as methyl, ethyl, butyl, etc.), aromatic groups (such as phenyl, indole, etc.) usually have a stronger PMR effect[1]. Moreover, tryptophan, which has an indole side chain, has a stronger PMR effect than phenylalanine, which has a benzyl side chain. Therefore, we chose tryptophan as the hydrophobic end of the amphiphilic molecule.
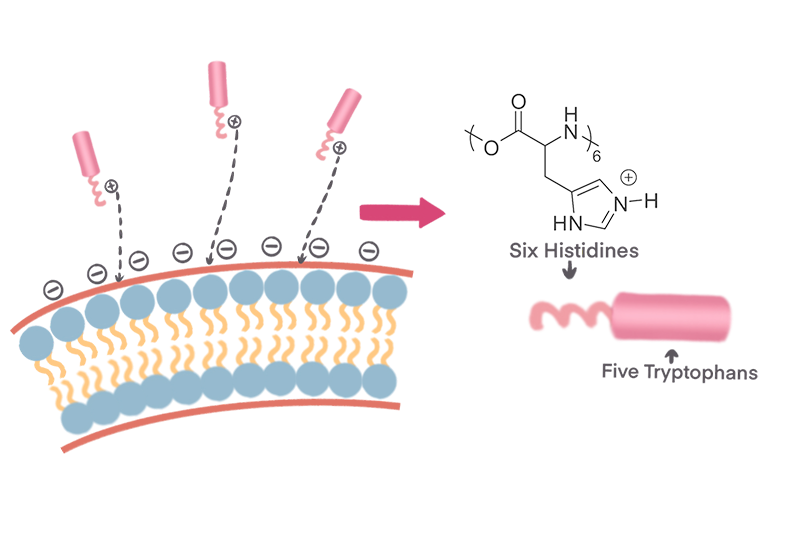
Another challenge is the choice of the cationic part, which has to consider the tumor microenvironment. In normal tissues, the extracellular pH value is strictly regulated to about 7.35-7.45, while in pathological conditions, such as local ischemia, inflammation and tumor, pH imbalance usually occurs. Due to the “Warburg effect”[2], tumor cells mainly rely on glycolysis, which produces a large amount of lactic acid. When a large amount of lactic acid accumulates in tumor cells, proton pumps transport H+ to the extracellular environment, resulting in an acidic extracellular environment[3] (pH=5.6~6.8). Therefore, weak acidity is not only a significant feature of the tumor microenvironment, but also an effective target for targeted delivery of anti-tumor drugs. The pKa of histidine is about 6.5, which is the only special amino acid among the 20 natural amino acids that constitute proteins whose pKa value is close to the pH value of the tumor microenvironment. Its structure contains an imidazole group[4], which is a strong nucleophilic group that can quickly provide and accept protons. In a neutral environment, histidine is uncharged and exhibits hydrophobic characteristics. And when pH<pKa, histidine is protonated and positively charged, and the electrostatic repulsion between molecules increases, making it easier for polymers to collapse. Based on the above principles, in the weakly acidic environment of the TME, our positively charged amphiphilic molecules (pTrp-pHis) will be more likely to be attracted by the negatively charged tumor cell membrane, thereby achieving relatively selective targeting and killing.
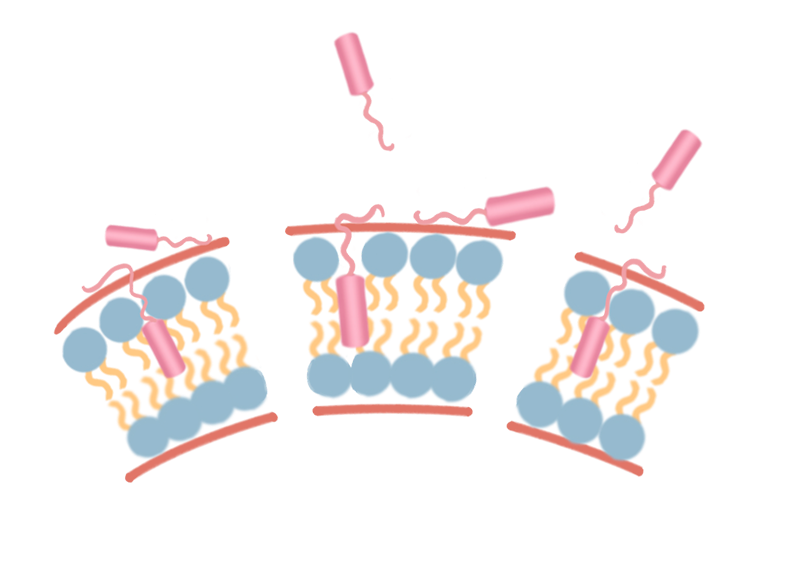
The high concentration of matrix metalloproteinase (MMP) in the tumor microenvironment is also an issue that cannot be ignored. Matrix metalloproteinase is a large family with similar structures and named for its need for metal ions such as Ca2+ and Zn2+ as cofactors. MMP can degrade almost all protein components in the extracellular matrix (ECM), destroying the histological barrier to tumor cell invasion. Its family member MMP-2 has long been associated with tumor invasion, metastasis and angiogenesis[5,6]. There is enough evidence to support that MMP-2 promotes tumor invasion and metastasis, especially solid malignant tumors such as liver cancer[7,8], lung cancer[9], stomach cancer[10], breast cancer[11] and so on. This indicates that a high MMP-2 condition in TME can be used to achieve relative selective targeting of drugs.
We designed this nanoparticle by integrating several major strategies: self-assembly, PEGylation, EPR effect, and stimulus responsiveness. Our aim was to develop a drug-free antineoplastic strategy for cancer therapy. We expected that our innovative treatment strategy would exhibit superior tumor toxicity and anti-tumor drug resistance. It would also possess unique abilities to induce cell death in diverse cancer cell phenotypes. In future studies, we plan to optimize the peptide design, perform enzyme cleavage response experiments, assess the anti-tumor activity of the peptide on various tumor cells, and evaluate its antibacterial activity.
By combining several major strategies: self-assembly, PEGylation, EPR effect, and stimulus responsiveness, we designed this nanoparticle. This enabled us to develop an innovative strategy to combat drug resistance in cancer therapy. We hypothesize that this strategy can address broader drug resistance challenges in treating bacterial and fungal infections.
[1] Liu M, Huang L, Zhang W, et al. A transistor-like pH-sensitive nanodetergent for selective cancer therapy[J]. Nature Nanotechnology, 2022, 17(5): 541-551.
[2] Abbas Z, Rehman S J N. An overview of cancer treatment modalities[J], 2018, 1: 139-157.
[3] Mookherjee N, Anderson M A, Haagsman H P, et al. Antimicrobial host defence peptides: functions and clinical potential[J], 2020, 19(5): 311-332.
[4] Iwasaki T, Tokuda Y, Kotake A, et al. Cellular uptake and in vivo distribution of polyhistidine peptides[J]. J Control Release, 2015, 210: 115-24.
[5] Karami Fath M, Babakhaniyan K, Zokaei M, et al. Anti-cancer peptide-based therapeutic strategies in solid tumors[J]. Cell Mol Biol Lett, 2022, 27(1): 33.
[6] Blanco-Míguez A, Gutiérrez-Jácome A, Pérez-Pérez M, et al. From amino acid sequence to bioactivity: The biomedical potential of antitumor peptides[J]. Protein Sci, 2016, 25(6): 1084-95.
[7] Wang L, Wang N, Zhang W, et al. Therapeutic peptides: current applications and future directions[J]. Signal Transduct Target Ther, 2022, 7(1): 48.
[8] Tan E, Wan T, Yu C, et al. ROS-responsive polypeptides for intracellular protein delivery and CRISPR/Cas9 gene editing[J]. Nano Today, 2022, 46: 101617.
[9] Gonzalez-Avila G, Sommer B, Mendoza-Posada D A, et al. Matrix metalloproteinases participation in the metastatic process and their diagnostic and therapeutic applications in cancer[J]. Crit Rev Oncol Hematol, 2019, 137: 57-83.
[10] Quintero-Fabián S, Arreola R, Becerril-Villanueva E, et al. Role of Matrix Metalloproteinases in Angiogenesis and Cancer[J]. Front Oncol, 2019, 9: 1370.
[11] Daniele A, Abbate I, Oakley C, et al. Clinical and prognostic role of matrix metalloproteinase-2, -9 and their inhibitors in breast cancer and liver diseases: A review[J]. Int J Biochem Cell Biol, 2016, 77(Pt A): 91-101.